1.技术原理与平台简介
拉曼光谱是一种基于分子非弹性散射的振动光谱技术。当入射光子与分子发生非弹性碰撞时,光子能量发生转移,拉曼位移对应于分子特定化学键或官能团的振动能级差,因此拉曼光谱可作为表征分子结构信息的“指纹”光谱。共聚焦拉曼显微镜在常规拉曼基础上引入共焦针孔设计,有效抑制焦平面以外的杂散光,可实现光学衍射极限的空间分辨(横向≤250 nm,纵向≤900 nm的空间分辨率,支持空气、液体及原位多场景测试。
本中心WITec alpha 300RAS共聚焦拉曼成像系统,采用光纤耦合超高通量光谱仪(UHTS300),配置多波长激光源(266 nm、532 nm),并集成原子力显微镜(AFM)与扫描近场光学显微镜(SNOM)功能模块,可在同一平台上实现化学成像、纳米形貌表征及突破衍射极限的近场光学成像,无需移动样品即可进行多维度关联测试。

依托上述硬件优势,该平台已具备能源材料、催化及高分子领域的多维表征能力。以下结合典型文献案例,展示其在不同研究场景中的应用潜力:
2. 电池材料
2.1. 电解液溶剂化结构解析
拉曼光谱因其对分子间相互作用的高度敏感性,成为研究电解液溶剂化结构的有效工具。通过与电化学原位池的联用,可进一步监测电极材料在充放电过程中的结构演变。在电解液研究中,拉曼光谱在800–1200 cm⁻¹范围内的C–O–C伸缩振动区可精确反映溶剂分子与Li⁺的配位状态变化。氟化醚类溶剂(如HFE)中氟原子的强吸电子效应削弱了醚氧原子的给电子能力,降低了溶剂分子与Li⁺的配位能力,形成弱溶剂化结构,这种结构促进了阴离子(如PF₆⁻、FSI⁻)进入Li⁺的第一溶剂化鞘层,在电化学还原过程中优先分解生成富含LiF的固体电解质界面(SEI)膜。拉曼光谱进一步证实,含氟化溶剂的电解液在锂金属表面形成的SEI膜具有更均匀的化学组成和更高的界面稳定性(J.Colloid Interface Sci.2025,702,138893)。
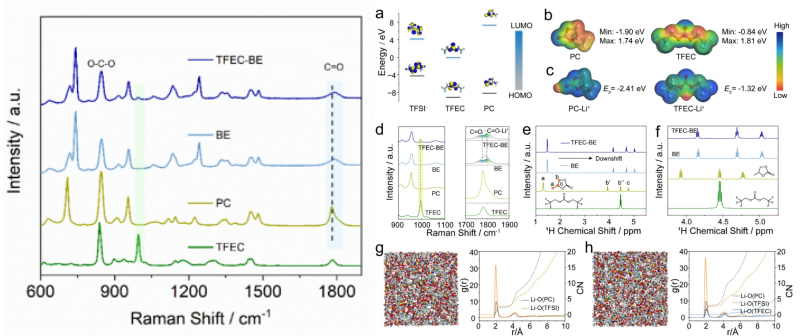
图1: 不同溶剂和电解质的拉曼光谱:(a)不同组分的HOMO-LUMO能级;(b)PC和TFEC的ESP密度分布;(c)Li⁺-PC和Li⁺-TFEC的结合能;(d)拉曼光谱以及(e)¹H NMR和(f)PC、TFEC和BE的放大NMR光谱,有/无TFEC;(g)BE和(h)TFEC-BE电解质的三维构象,以及对应的径向分布函数和配位数(CN)。
2.2 电极充放电过程监测
在负极材料结构研究方面,拉曼光谱对碳材料中sp²/sp³杂化比例变化高度敏感性,使其成为追踪碳骨架结构演变的关键手段。以钠离子电池硬碳负极为例,D峰(~1350 cm⁻¹,无序sp²碳)的与G峰(~1580 cm⁻¹,石墨化sp²碳)的强度比(ID/IG)随碳化温度的变化,精确反映了从无序结构到伪石墨结构、再到类石墨结构的转变过程。结合XRD与SAXS数据,首次从碳骨架振动的角度建立了伪石墨相变与闭孔形成的直接关联。原位拉曼光谱在放电过程中实时捕获了G峰的红移与强度衰减,并在~1450 cm⁻¹处观测到钠-碳化合物(Na-C)的振动峰,这一结果为Na⁺嵌入碳层的行为提供了直接光谱证据,支持了“吸附-嵌入-填充”储钠模型的建立。上述原位实验采用自制的原位电化学拉曼池(石英光学窗口),实现了充放电过程中电极表面拉曼信号的实时采集(J. Colloid Interface Sci.2025,702,138855)。
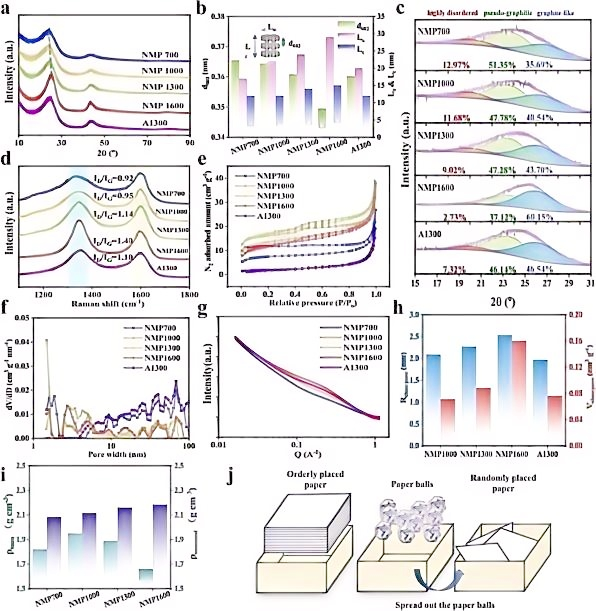
图2: (a)XRD图谱;(b)微晶结构数据;(c)XRD拟合曲线;(d)拉曼光谱;(e)N₂吸附-脱附等温线;(f)孔径分布;(g)小角X射线散射图谱;(h)闭孔信息;(i)样品真密度和骨架密度;(j)“纸板箱”理论示意图。
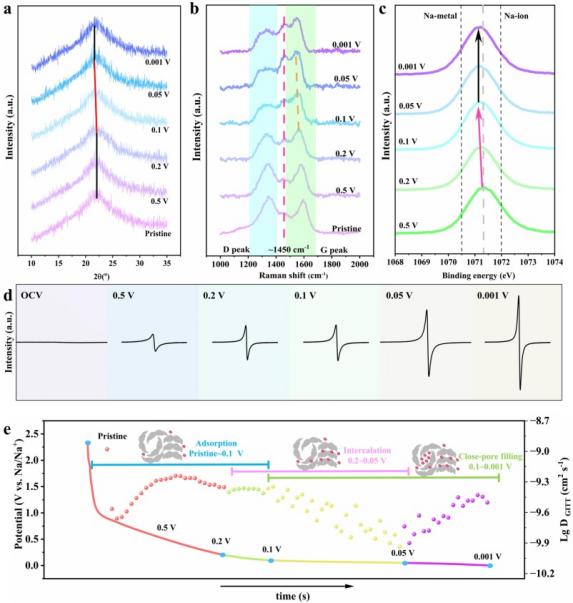
图3: (a)原位XRD光谱;(b)原位拉曼光谱;(c)非原位XPS光谱;(d)NMP1300电极在不同放电阶段的EPR光谱;(e)储钠模型示意图。
上述案例表明,拉曼光谱在电池研究中兼具““静态分析与动态检测功能:既可解析电解液溶剂化结构的静态化学环境,又能实时追踪电极材料在充放电过程中结构演变与相变过程,从而建立分子振动光谱与电化学性能之间的关联,为电池界面工程和电极材料设计提供依据。
拉曼光谱在催化研究中兼具静态结构解析与动态过程监测功能。通过耦合原位反应池,可在反应条件下实时监测催化剂表面吸附物种的演变;采用深紫外(266 nm)激发则可有效抑制沸石分子筛等载体的荧光干扰,进而获取金属–配体配位结构的振动信息。
3.1 电催化CO₂还原
本研究利用原位共聚焦拉曼光谱(配备三电极原位电化学池),在–0.6V至–1.4V(vs. RHE)电位范围内监测了Cd-Sb烧绿石氧化物(Cd₂Sb₂O₇)催化剂表面的物种演变。100–1800 cm⁻¹范围内随外加电位依赖拉曼光谱清晰揭示了关键反应中间体的动态行为:~703c m⁻¹处归属于CO₂⁻的特征峰证实了催化剂对CO₂分子的有效活化;~1640 cm⁻¹处COOH中间体的特征峰随电位负移先增强后减弱,直接反映了关键中间体的动态生成与转化过程;~267 cm⁻¹处*CO在Cd位点上的受限旋转振动峰,以及~624 cm⁻¹处Sb–O振动峰随电位的响应,共同揭示了Cd活性位点与Sb位点局域配位环境在反应中的协同作用。值得注意的是,~2000 cm⁻¹附近未检测到CO特征峰,表明反应生成的CO能够快速脱附,避免了活性位点毒化(Chem. Eng.J.2026,174919)。
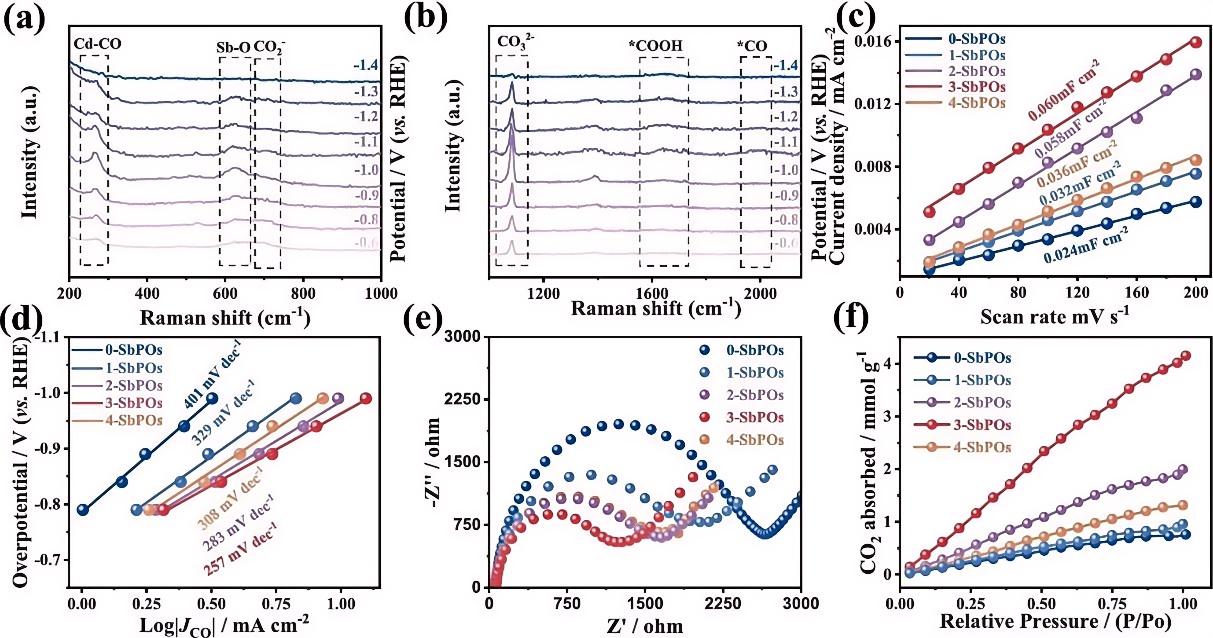
图4: (a,b)3-SbPOs在不同电位下的原位拉曼光谱;(c)化学分析电子能谱(ESCA)图;(d)产CO的Tafel图;(e)x-SbPOs的Nyquist图及拟合曲线;(f)CO₂吸附等温线。
3.2 热催化CO氧化
本研究利用原位共聚焦拉曼光谱结合紧凑型高温原位反应池,在150°C、流动CO/O₂气氛下对Cu/CeO₂催化剂进行了实时光谱采集。拉曼光谱中CeO₂的F₂g特征峰(~465 cm⁻¹)在反应气氛切换和温度变化过程中发生可逆的位移与展宽,这一现象源于CeO₂载体表面氧空位浓度的动态变化:氧空位增多时晶格膨胀,F₂g峰红移;氧空位被填充时晶格收缩,F₂g峰恢复至初始位置。与此同时,Cu–O相关振动模式(~290 cm⁻¹、~630 cm⁻¹)的强度与峰位变化揭示了Cu物种在反应过程中的价态演变(Cu⁺ ↔ Cu²⁺),将其与气相色谱记录的CO转化率变化进行时序关联分析,发现Cu⁺/Cu²⁺的动态平衡与催化活性的振荡行为之间存在定量对应关系。上述结果从分子振动光谱层面为明确负载型铜基催化剂CO氧化的活性位点本质及反应路径提供了实验依据(Catal.Sci.Technol.2025,15,3581)。
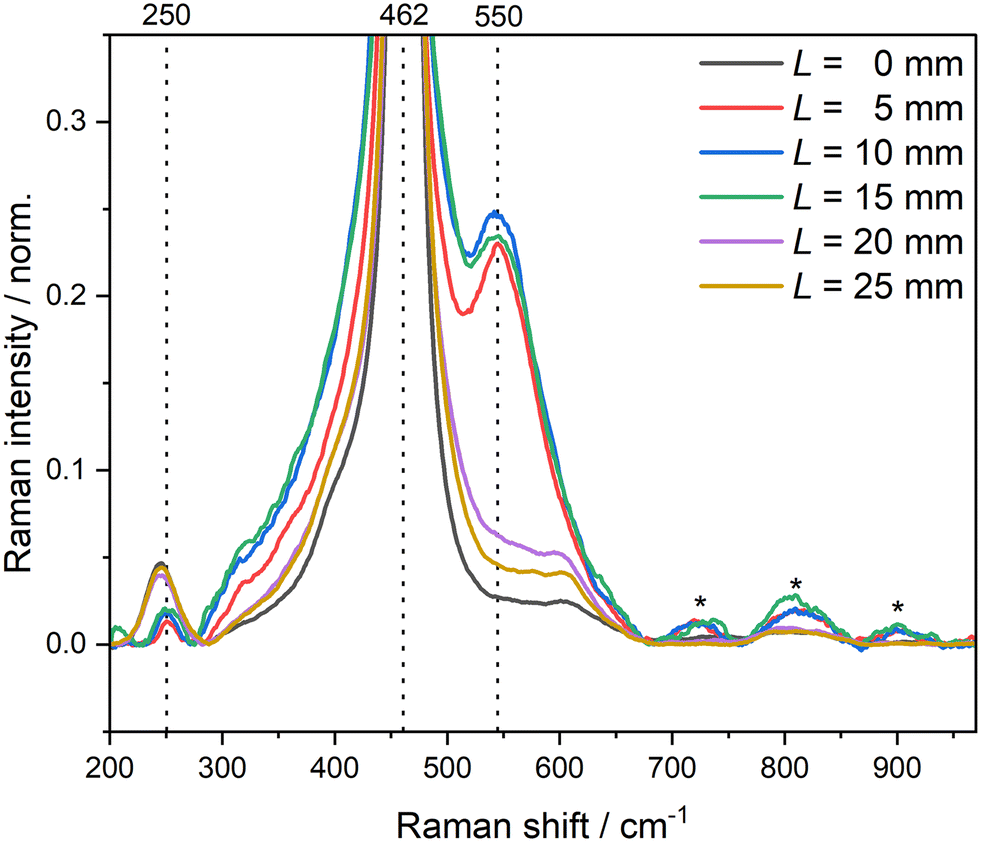
图5: 0.38wt% Cu/CeO₂催化剂在CO氧化反应过程中,于632.8nm激发下采集的拉曼光谱(以F₂g峰归一化)。*标记为仪器假峰。
3.3 深紫外拉曼
本研究采用深紫外拉曼光谱(266 nm激发)对金属–沸石催化剂合成过程中的配位结构进行了追踪。深紫外激发可有效避免沸石骨架的荧光干扰,可直接获取金属–配体的振动信息。研究系统揭示了多氨基策略(MAE)中1,3-二氨基丙烷与Cu²⁺物种之间的配位相互作用及其对Cu活性位点形成的调控机制。拉曼光谱首先确认了MAE-MOR样品中1,3-二氨基丙烷以完整分子形式被引入MOR孔道,并观测到–NH₃⁺物种的特征谱带(~1035 cm⁻¹),证明其一端氨基通过质子化锚定于分子筛骨架,而另一端氨基保持游离状态。进一步对比发现,传统离子交换法制备的H-CuM-3中Cu²⁺相关谱带位于166 cm⁻¹,归属于Cu²⁺与骨架氧原子的配位;而MAE-CuM-1中该谱带红移至157 cm⁻¹,直接证实了MAE策略中Cu²⁺与–NH₂基团之间形成了独特的配位键,而非传统离子交换中Cu²⁺与骨架酸位点的简单置换。该配位相互作用不仅提高了Cu物种的负载量,还抑制了Cu物种在焙烧过程中的团聚,同时多氨基结构在焙烧过程中分阶段释放NH₃,实现Cu物种的原位自还原,获得高比例Cu⁺与Cu⁰共存的高分散活性位点(Chem.Eng.J.2025,525,170712)。
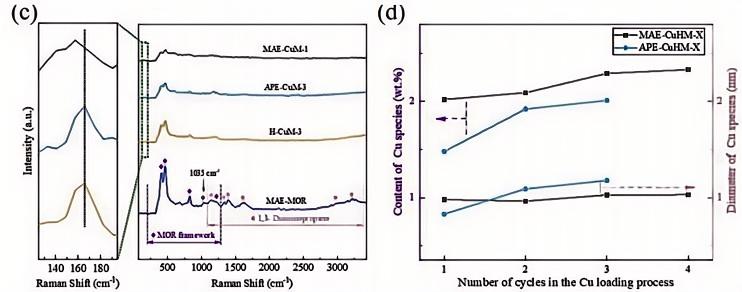
图6:(a)MAE-MOR、H-CuM-3、APE-CuM-3和MAE-CuM-1的拉曼光谱;(b)所有自还原CuHM催化剂中Cu物种含量/粒径与Cu负载循环次数的关系。
上述研究从电催化中间体演变、热催化活性位点动态变化以及催化剂活性位点形成机制三个维度,展示了拉曼光谱在催化研究中的多维表征优势。
4. 高分子材料:PAN预氧化纤维结构均一性评价
预氧化纤维(OFs)的皮–芯结构是决定碳纤维最终力学性能的关键因素之一。本研究采用共聚焦拉曼光谱二维扫描成像技术,结合光学显微镜与扫描电子显微镜(SEM),系统分析了不同密度(1.29–1.43 g·cm⁻³)PAN基预氧化纤维的径向结构不均匀性及其演变规律。拉曼光谱结果显示,原丝(PFs)在2244 cm⁻¹处呈现C≡N伸缩振动峰,在2800–3000 cm⁻¹范围内呈现多重C–H伸缩振动峰;预氧化后这些信号基本消失,同时出现D峰(~1350 cm⁻¹,无序结构A₁g振动模式)和G峰(~1600 cm⁻¹,有序石墨结构E₂g振动模式),表明sp³杂化碳向sp²杂化碳的转化。通过“66×66”点阵的拉曼面扫描(覆盖20×20 μm区域,空间分辨率约300 nm),获得AG/AD比值(G峰与D峰积分面积比)的空间分布图像。所有OFs均呈现芯部暗(AG/AD较低)而皮层亮(AG/AD较高)的分布规律,表明皮层预氧化程度高于芯部,且该径向不均匀性随密度增加而加剧。该工作从分子振动光谱角度为预氧化纤维皮层-芯层结构的形成机制及密度调控提供了直接实验依据,并指出密度范围为1.35–1.37 g·cm⁻³的OFs是制备高质量碳纤维的最佳密度范围(J.Appl.Polym.Sci.2025)。
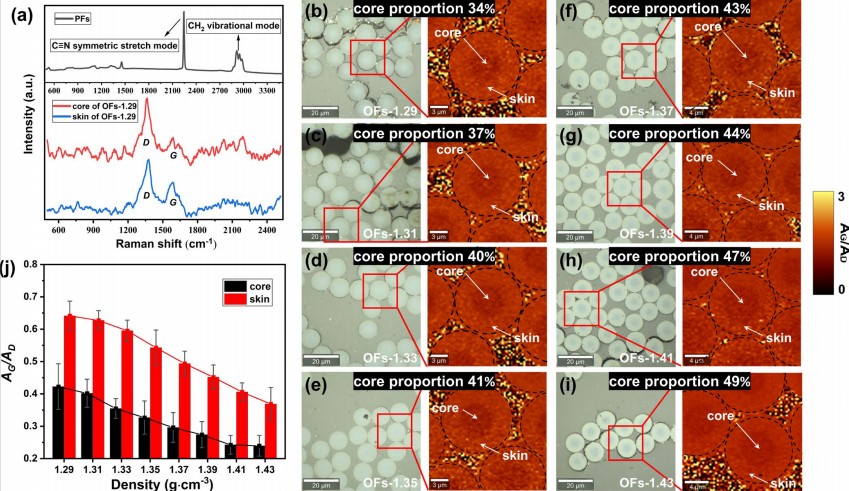
图7: (a)PFs和OFs的拉曼光谱;(b–i)PFs和OFs的光学显微镜图像及AG/AD比值面扫描成像图;(j)皮层与芯部AG/AD比值与OFs密度的关系。
共聚焦拉曼光谱技术兼具非破坏性、免标记、高空间分辨率和多波长激发等优势,适用于能源材料与催化体系的结构表征与动态过程研究。在电池研究领域,拉曼光谱既可解析电解液溶剂化结构与界面化学组成,也可追踪电极材料碳骨架的与充放电过程中的动态结构演变。在催化研究领域,结合原位电化学池或高温气氛反应池,可实时捕获反应中间体、活性位点价态变化及载体氧空位动态等关键信息。在聚合物及复合材料研究领域,面扫描成像模式可直观呈现径向结构与界面组分的空间分布特征。该技术可与扫描电镜-能谱(SEM-EDS)、X射线光电子能谱(XPS)、透射电镜(TEM)、原子力显微镜(AFM)等多种表征方法形成互补,构建从分子振动到宏观性能的完整表征体系,为高性能能源材料与催化材料的研发提供关键技术支撑。
附件下载: